Bioinformatics
Our laboratory is engaged in the development and application of bioinformatics tools used in a wide range of biomedical studies.
Protein Disulfide Engineering
Disulfides are naturally occurring bonds that provide stability to many proteins. Disulfides also restrict motion in protein regions that are bonded together. It is advantageous to artificially increase the stability of some proteins, for example to improve the stability of industrial enzymes or peptide-based pharmaceuticals. Additionally, to assist studies of protein dynamics and function, it is often desirable to insert a disulfide bond to restrict motion or alter enzymatic activity. Disulfide engineering is the directed design of novel disulfide bonds into proteins. We have developed algorithms and software to enable investigators to effectively utilize this biotechnological method by accurately predicting locations where disulfide bonds can successfully form in a protein.
Disulfides are naturally occurring bonds that provide stability to many proteins. Disulfides also restrict motion in protein regions that are bonded together. It is advantageous to artificially increase the stability of some proteins, for example to improve the stability of industrial enzymes or peptide-based pharmaceuticals. Additionally, to assist studies of protein dynamics and function, it is often desirable to insert a disulfide bond to restrict motion or alter enzymatic activity. Disulfide engineering is the directed design of novel disulfide bonds into proteins. We have developed algorithms and software to enable investigators to effectively utilize this biotechnological method by accurately predicting locations where disulfide bonds can successfully form in a protein.
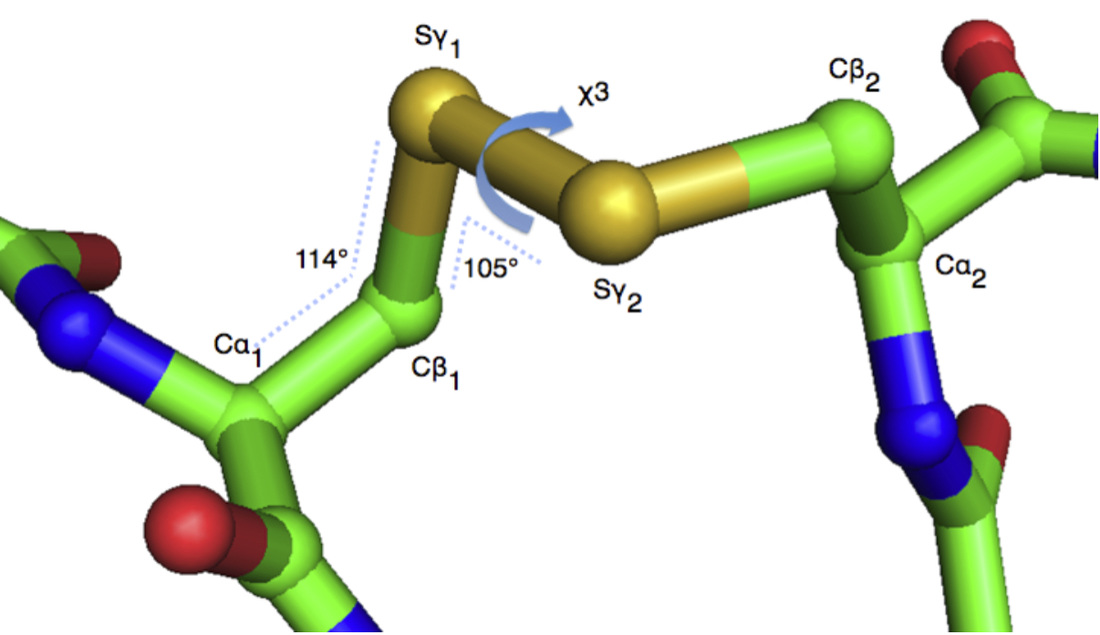
Disulfide bond geometry. From Dombkowski et al., FEBS Letters, 2014, Vol. 588, Issue 2, p 206-212
|
To create a new disulfide bond, two amino acids (residues) in close spatial proximity must be mutated to cysteines. However, spatial proximity alone is not an efficient selection criteria for designing novel disulfide bonds. Cysteines involved in disulfides bonds conform to strict structural constraints. Therefore, we have developed bioinformatics tools to select residues in a target protein where mutation to cysteines will likely form a disulfide. Our computational algorithms are based on analysis of the geometry of hundreds of naturally occurring disulfide bonds. We have developed very accurate disulfide prediction methods, and our computational tools have been used in many published studies, including reports in Nature, Cell, and Science. We have licensed this technology to pharmaceutical firms.
|
Disulfide by Design used to expedite the search for a COVID-19 vaccine.
Four international research groups used DbD2 to assist in development of multi-epitope peptide vaccines for the SARS-CoV-2 virus:
- Ismail, S., S. Ahmad, and S.S. Azam, Immuno-informatics Characterization SARS-CoV-2 Spike Glycoprotein for Prioritization of Epitope based Multivalent Peptide Vaccine.bioRxiv, 2020: p. 2020.04.05.026005.
- Sarkar, B., et al., The Essential Facts of Wuhan Novel Coronavirus Outbreak in China and Epitope-based Vaccine Designing against 2019-nCoV.bioRxiv, 2020: p. 2020.02.05.935072.
- Rehman, H.M.M., M.U.; Saleem, M.; Froeyen, M.; Ahmad, S.; Gul, R.; Aslam, M.S.; Sajjad, M.; Bhinder, M.A., A Putative Prophylactic Solution for COVID-19: Development of Novel Multiepitope Vaccine Candidate against SARS‐COV‐2 by Comprehensive Immunoinformatic and Molecular Modelling Approach. Preprints, 2020.
- Nazneen Akhand, M.R., et al., Genome based Evolutionary study of SARS-CoV-2 towards the Prediction of Epitope Based Chimeric Vaccine.bioRxiv, 2020: p. 2020.04.15.036285.
Tools for Functional Genomics
|
Better Bunny: Augmented Annotation and Orthologue Analysis for Oryctolagus cuniculus.
The rabbit is an important model organism used in many biomedical research studies. However, functional genomics studies using the rabbit have been impeded by sparse annotation of the genome, thus prohibiting extensive functional analysis of gene sets derived from whole-genome experiments. To address this shortcoming, we developed a web-based application that provides extensive functional annotation for rabbit genes, through orthologous relationships and integration of several external databases. |

|

miR-AT!: miRNA Combinatorial Analysis of Targets
miR-AT! is a computational tool for the identification of transcripts that are targets of a list of input miRNAs. Targets are identified using the Sanger MicroCosm Targets database, and the output provides a list of all computationally predicted targets, the number of sites in each transcript and the cumulative score. Filters can be applied to restrict target site selection. Links for each transcript in the output provide additional information from the NCBI and Ensembl databases, as well as miR target site locations. miR-AT! also enables the automated submission of output target lists to DAVID for functional annotation of ontologies and pathways.
miR-AT! is a computational tool for the identification of transcripts that are targets of a list of input miRNAs. Targets are identified using the Sanger MicroCosm Targets database, and the output provides a list of all computationally predicted targets, the number of sites in each transcript and the cumulative score. Filters can be applied to restrict target site selection. Links for each transcript in the output provide additional information from the NCBI and Ensembl databases, as well as miR target site locations. miR-AT! also enables the automated submission of output target lists to DAVID for functional annotation of ontologies and pathways.
